O.o!
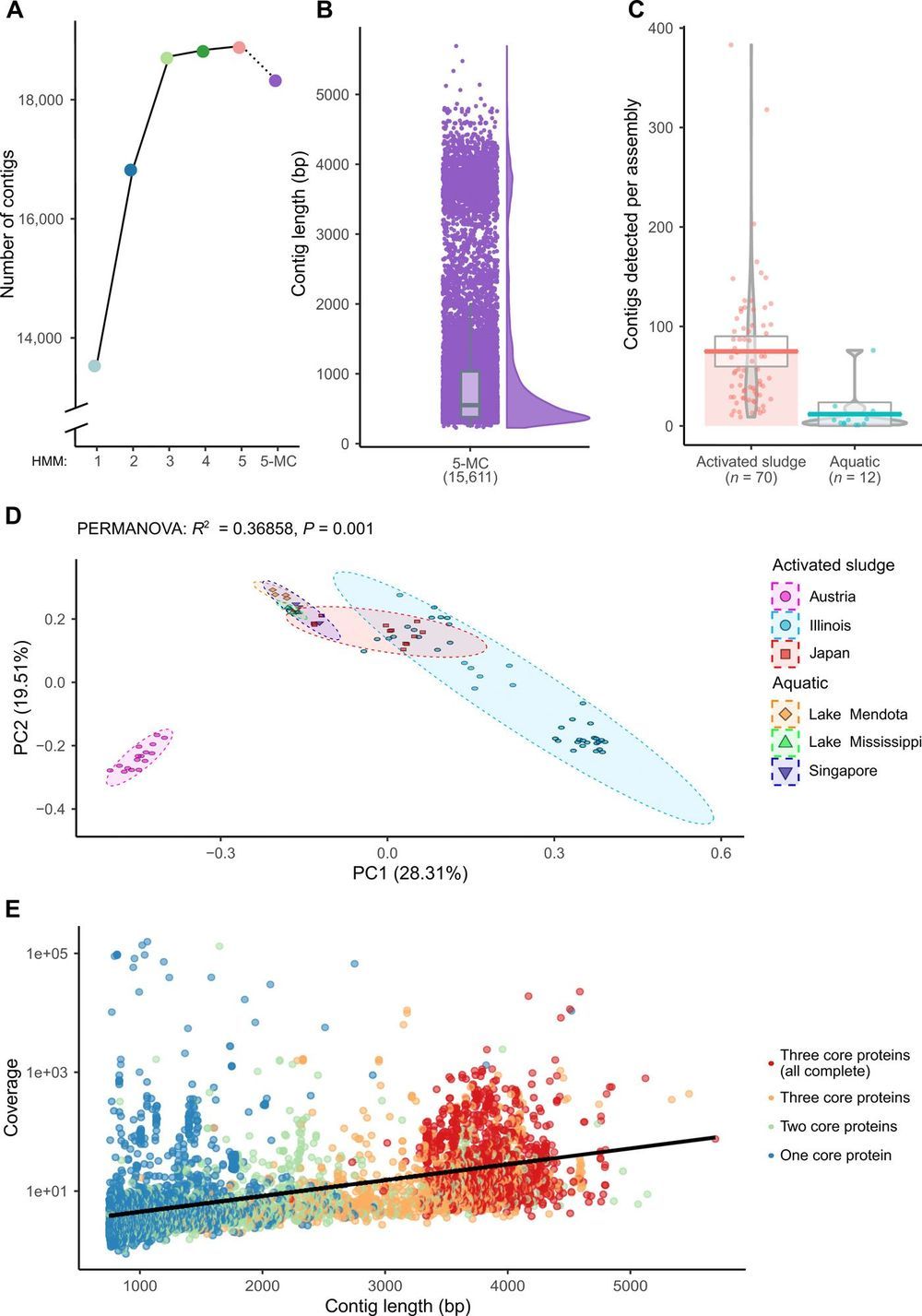
The first sequenced genome was that of the 3569-nucleotide single-stranded RNA (ssRNA) bacteriophage MS2. Despite the recent accumulation of vast amounts of DNA and RNA sequence data, only 12 representative ssRNA phage genome sequences are available from the NCBI Genome database (June 2019). The difficulty in detecting RNA phages in metagenomic datasets raises questions as to their abundance, taxonomic structure, and ecological importance. In this study, we iteratively applied profile hidden Markov models to detect conserved ssRNA phage proteins in 82 publicly available metatranscriptomic datasets generated from activated sludge and aquatic environments. We identified 15,611 nonredundant ssRNA phage sequences, including 1015 near-complete genomes. This expansion in the number of known sequences enabled us to complete a phylogenetic assessment of both sequences identified in this study and known ssRNA phage genomes. Our expansion of these viruses from two environments suggests that they have been overlooked within microbiome studies.
Viruses, particularly bacteriophages targeting prokaryotes, are the most diverse biological entities in the biosphere (1, 2). Currently, there are 11,489 genome sequences available in the NCBI (National Center for Biotechnology Information) Viral RefSeq database (version 94). The vast majority of known phage have a double-stranded DNA (dsDNA) genome (3, 4). Recent metagenomic analysis of 145 marine virome sampling sites identified 195,728 DNA viral populations, highlighting that only a fraction of Earth’s viral diversity has been characterized (5). An additional expansion of known phage populations by Roux et al. (6) revealed that not only dsDNA phages but also single-stranded DNA Inoviridae are far more diverse than previously considered. The rapid expansion in viral discovery through metagenomics is enabling a greater understanding of their roles within environments and their evolutionary relationships, which is subsequently causing a revolution in phage taxonomy (7).
Despite the identification of single-stranded RNA (ssRNA) phages over 50 years ago (8), there are few representative sequences available. The International Committee on Taxonomy of Viruses (ICTV) has currently categorized approximately 5500 viruses (9). Yet, their classification only applies to 25 ssRNA phage sequences (complete or partial) across two genera, Levivirus and Allolevivirus, and an additional 32 sequences unclassified below a family taxonomic rank (10). Historically, methods for classifying Leviviridae depended on molecular weight, density, sedimentation, and serological cross-reactivity (11). A subsequent classification method separated the two genera, with the Alloleviviruses containing a fourth unique gene predicted to encode a lysin (12). Recently, an analysis of the evolution origin of all currently known RNA viruses by Wolf et al. (13) suggested that ssRNA phages may actually be two distinct lineages, which they termed Leviviridae and “Levi-like” viruses.