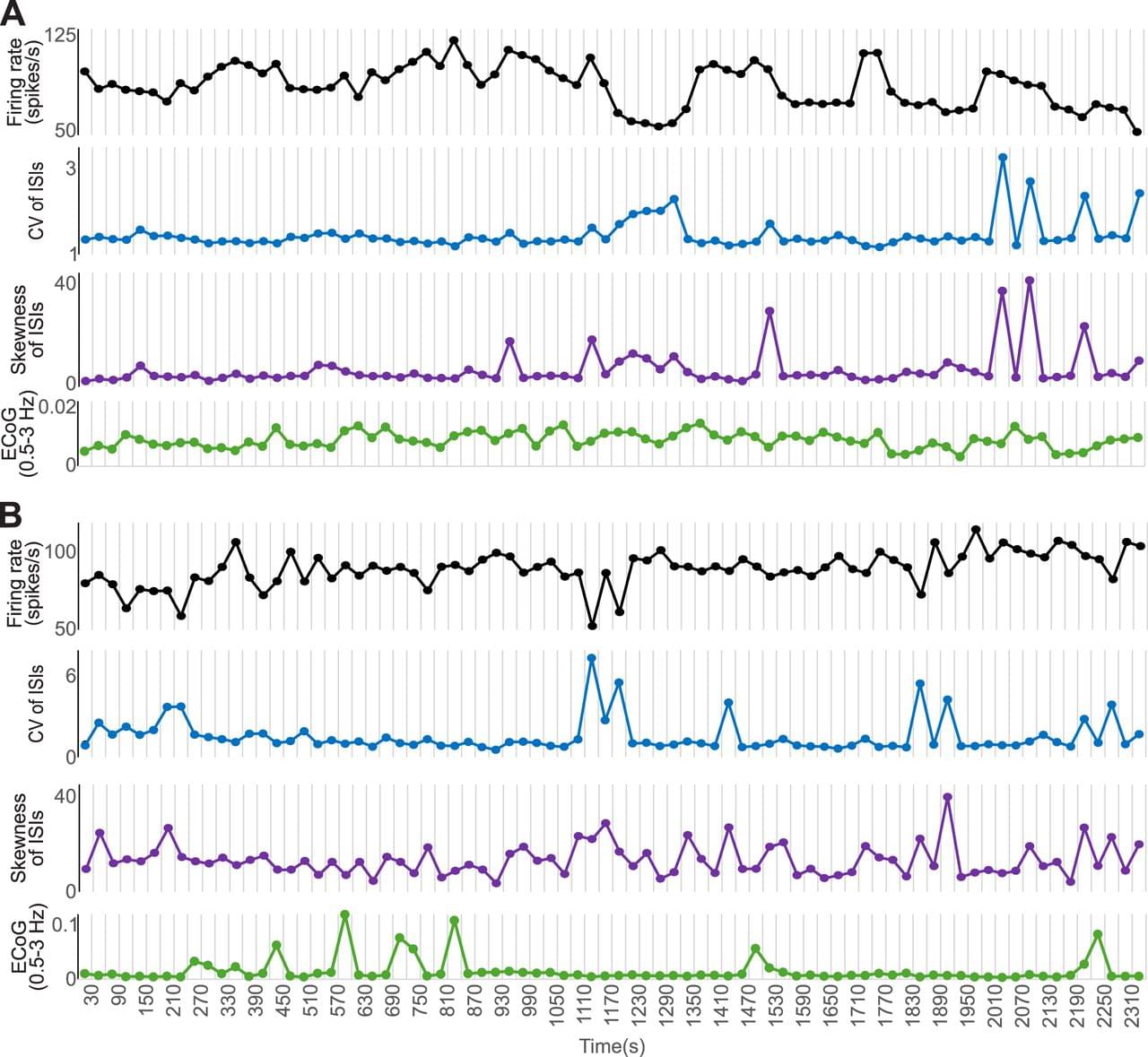
The first report of in vivo extracellular recordings in the external segment of the globus pallidus (GPe) of awake monkeys described that most GPe neurons show high-frequency spiking activity, interspersed with pauses (high-frequency discharge with pauses, HFD-P), while a smaller proportion was said to show low-frequency discharge with bursts (LFD-B; DeLong, 1971). Similar patterns of pallidal discharge have been demonstrated by other authors, both in primates (Katabi et al., 2022) and rodents (Bugaysen et al., 2010; Benhamou et al., 2012).
There is evidence, however, that the HFD-P and LFD-B subtypes of GPe neurons are only the most recognizable extremes of a continuous spectrum of properties of GPe neurons. This view is supported by in vivo and in vitro recordings in rodents which found that the firing properties of the population of GPe neurons distribute along a continuum, with specific cells firing within more limited boundaries of firing rates and patterns (Abdi et al., 2015; Cui et al., 2021). Furthermore, observations in rodents showed that GPe neurons display a wide range of firing rates and patterns (Deister et al., 2013). The firing pattern heterogeneity in in vivo recordings may arise, at least in part, due to shifts in firing behavior of the same neurons, as has been reported in rodent studies (Deister et al., 2013). Such variations in firing patterns may only be detectable when recordings extend over long time periods (Elias et al., 2008).
The loss of nigrostriatal dopamine fibers associated with parkinsonism induces multiple alterations in GPe, where neuronal firing becomes slower, may show more frequent bursts, and becomes more synchronized (Galvan et al., 2015; Courtney et al., 2023). However, the stability of firing patterns of GPe neurons in the parkinsonian state has not yet been investigated.