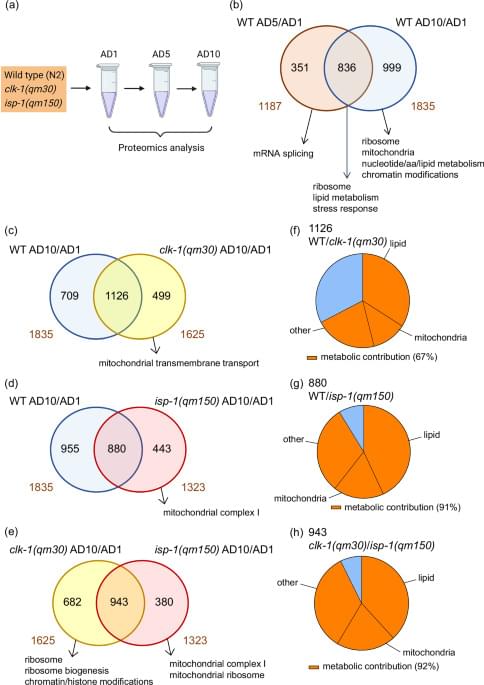
Mitochondrial decline impairs late-life metabolic plasticity. Using nematodes and human data, this study identifies reduced phosphatidylcholine synthesis as a natural trigger of mitochondrial dysfunction during aging.
Dr. Fahy is the Vice President and Chief Scientific Officer at 21st Century Medicine, Inc, and has co-founded Intervene Immune, a company developing clinical methods to reverse immune system aging. He was the 2022–2023 president of the Society for Cryobiology. Dr. Fahy is the lead author of a recent paper, “Ultrastructural and Histological Cryopreservation of Mammalian Brains by Vitrification” – the main topic of our conversation.
In December of 2014, I worked with Dr. Fahy to cryopreserve Dr. Stephen Coles under special conditions, with his permission to extract brain samples and test them for preservation quality. We did not know what the results would be. If bad, that would be discouraging for cryonics. In fact, the results were excellent, as Dr. Fahy details.
We discuss the Coles case and the results of the cerebral cortical biopsy. The paper includes results from rabbit brains. We also discuss the relative resilience of the brain compared to other organs when it comes to fracturing; how cryoprotectants prevent ice formation even when the blood-brain barrier remains closed; whether biostasis organizations should be using blood-brain barrier opening agents; Dr. Fahy’s thoughts about chemical preservation and the role of a combination of cryo an chemo, known as aldehyde-stabilized cryopreservation (ASC), and more.
Hi all, you should check out this human enhancement game created by Anton Kulaga and his team! You’ll learn about various genetic factors with potential for increasing human capabilities. The content is all rooted in scientific literature! Once you design a character, you can also get a 3D printed shape sent to you which is influenced by your choices.
Build your post-human character from real genes — tardigrade radiation shields, naked-mole-rat cancer resistance, Greenland shark longevity — backed by scientific evidence tiers and real citations. Spend enhancement credits and 3D-print the result.
What if aging isn’t just “getting older” but the gradual breakdown of the biological systems protecting our DNA? Scientists are now exploring therapies designed to directly lengthen telomeres, the protective caps linked to cellular aging itself. The future of longevity may be far more programmable than we thought.
Heart disease still kills nearly 20 MILLION people every year worldwide — roughly 1 person every 1.5 seconds. — But what if medicine could move beyond simply slowing plaque buildup…and actually REMOVE toxic oxidized cholesterol from arteries? — Dr. Matthew “Oki” O’Connor, CEO and Co-Founder, Cyclarity Therapeutics.
In the time it will take you watch this episode, over 2,000 people around the world will die from diseases driven by arterial plaque. But what if we could actually remove the toxic cholesterol already trapped inside arteries?
Today we’re diving into one of the biggest unsolved problems in medicine and aging: how do you actually remove arterial plaque instead of merely slowing its progression?
Cardiovascular disease remains the world’s leading killer, despite decades of statins, anti-inflammatory drugs, and newer RNA-based therapies. Most existing treatments help manage cholesterol and reduce risk, but very few directly target the toxic debris already embedded inside plaques.
But what if we could literally extract some of the most dangerous oxidized cholesterol molecules from the body?
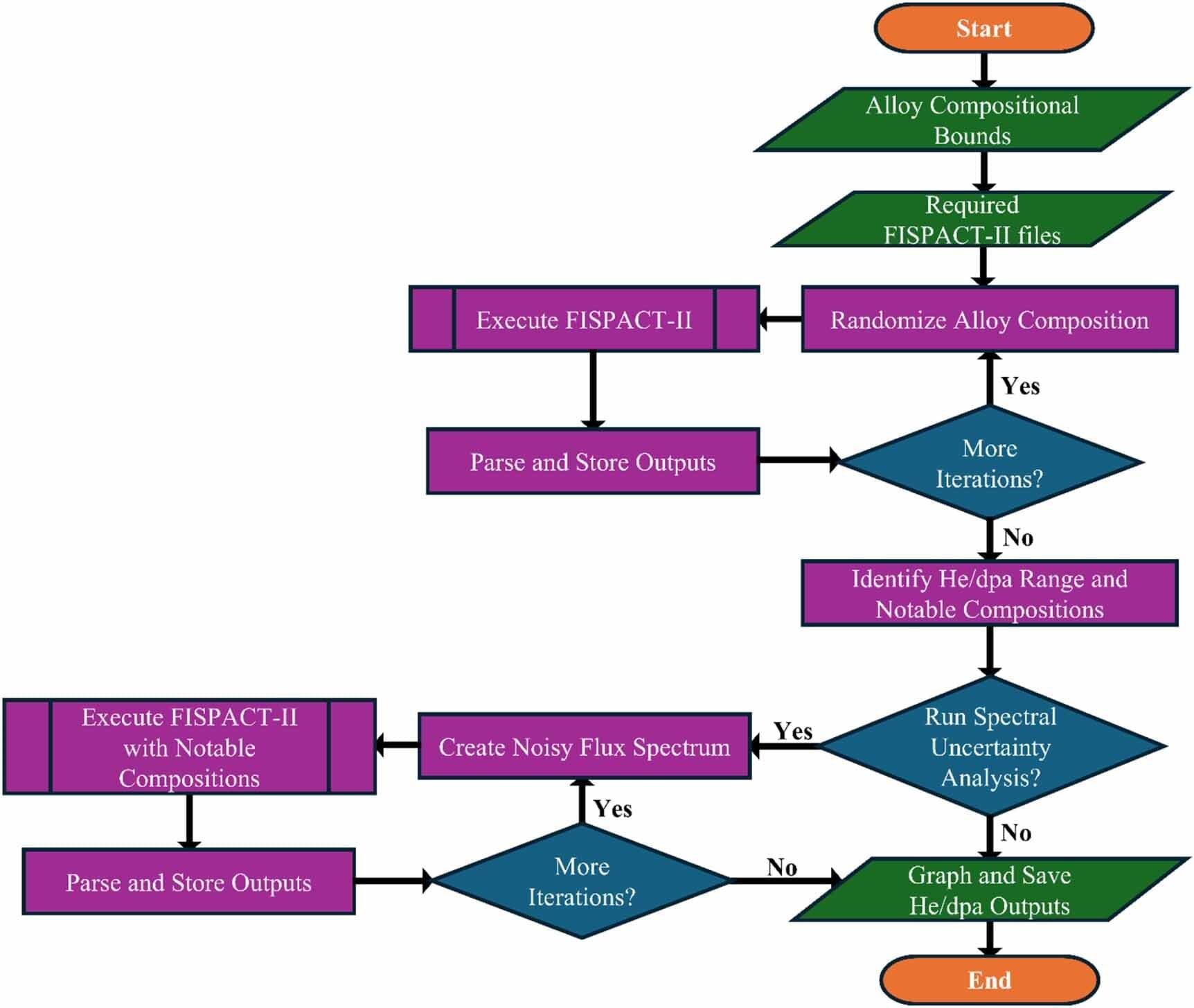
Standardizing calculations of the helium byproducts generated in advanced fission and fusion energy system materials can increase reactor safety and longevity, according to a study led by University of Michigan Engineering with collaborators at Oak Ridge National Laboratory and its management contractor UT-Battelle.
Through a series of simulations, the researchers found that modeling assumptions and key alloy elements—like carbon, nitrogen and nickel—significantly influence helium generation predictions. If left unaddressed, excess helium in real-world reactors could lead to faster component failure as materials swell and become brittle.
“If used, our reporting methods will improve the experimental and modeling fidelity of the nuclear materials databases being generated both domestically and internationally, driving the rapid deployment of advanced nuclear,” said Kevin Field, a professor of nuclear engineering and radiological sciences at U-M and corresponding author of the study published in the Journal of Physics: Energy.
What happens when biology starts behaving less like fate and more like software?
Some of the most controversial ideas in longevity research sound unbelievable at first… until the data starts forcing better questions.
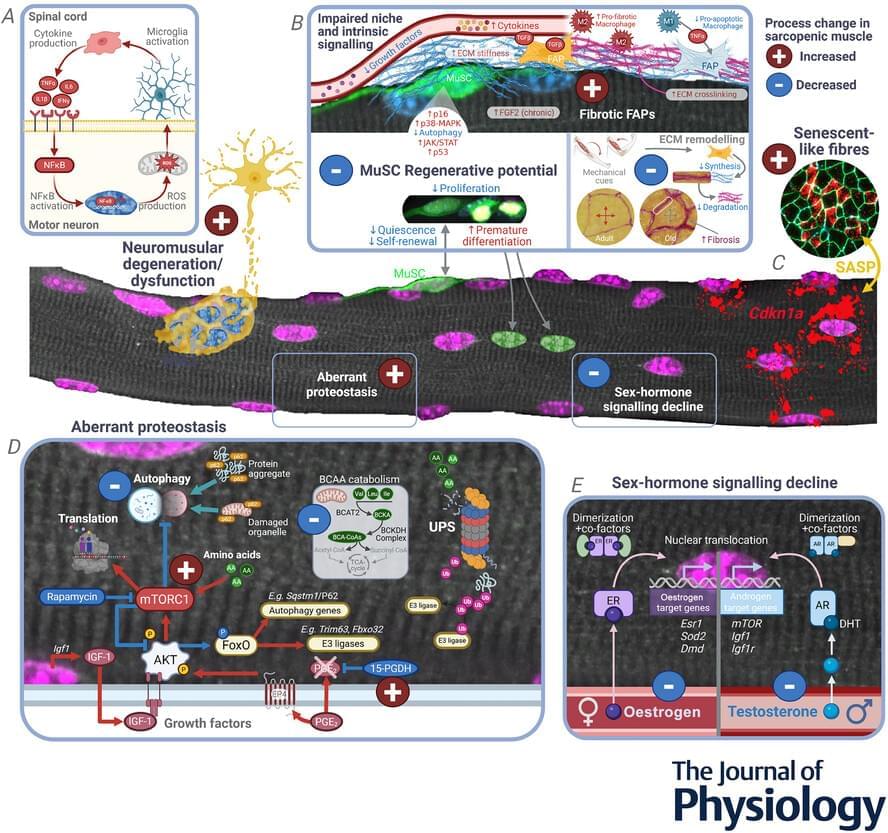
Aging human breast atlas reveals cancer susceptibility
The team used advanced imagining techniques to analyse breast tissue from more than 500 women aged 15 to 86 years old. The tissue included biopsies taken from women for non-cancer-related reasons.
Combining these images with details of the hormone receptors and immune cells present, as well as the tissue architecture, the researchers were able to map how breast tissue changes over time in unprecedented detail. Their findings point to reasons why breast cancer risk increases with age and why tumors in younger women differ biologically.
The author added: “Our map revealed that as women age, their breast tissue goes through major changes, with the most dramatic changes occurring at menopause. There are changes, too, during their twenties, possibly linked to pregnancy and childbirth, but these are far less pronounced.”
The map revealed that all types of cells become fewer in number and divide far less often. Milk-producing structures known as lobules shrink or disappear, while the ducts that that carry milk become relatively more common, with the supporting layer around them becoming thicker. Fat cells increase while blood vessels decrease.
Meanwhile, changes occur in the immune environment. Younger breasts have more B cells and active T cells, which helps them identify and kill cancer cells. As tissue ages, these types of cells decline in number, replaced by other types of immune cell that indicate a more inflammatory and potentially less protective immune environment. ScienceMission sciencenewshighlights.

Researchers at Bar-Ilan University have successfully restored youthful patterns of DNA organization in the livers of old mice, reversing key molecular features associated with aging. The study, published in Nature Communications, identifies the protein SIRT6 as a powerful protector against age-related breakdown in chromatin, the complex system that packages DNA and controls how genes are switched on and off.
The findings suggest that aging is not simply a passive process of wear and tear, but may be driven in part by reversible changes in the way DNA is organized inside cells.
DNA inside cells is tightly folded and packaged into chromatin, a structure that acts like a biological control system for gene activity. Using advanced tools to study DNA organization and gene activity, the researchers examined multiple molecular changes in the livers of young and old mice. What they discovered was dramatic: aging disrupts chromatin architecture in the liver, causing inflammatory pathways to become overactive while weakening the metabolic programs that define healthy liver tissue.