Fungal infections present persistent therapeutic challenges in immunocompromised populations, including individuals with acquired immunodeficiency syndrome (AIDS), organ transplant recipients receiving immunosuppressive therapy, long-term hospitalized patients, patients with cancer, and those receiving immunomodulatory agents.1 These infections demonstrate remarkable recalcitrance to conventional therapies, compounded by fungal adaptability to environmental stresses, the emergence of drug-resistant strains, and the limited availability of clinically available antifungal agents.2 Systemic fungemia has alarmingly high mortality rates, accounting for approximately 1.5 million annual deaths worldwide, a burden comparable to AIDS-and tuberculosis-related mortality.3 Candida albicans is the most frequently isolated fungal pathogen in clinical settings. Despite therapeutic advances, invasive candidiasis persists with mortality rates exceeding 40%,4 underscoring the urgent need to elucidate host immune mechanisms against fungal pathogens.
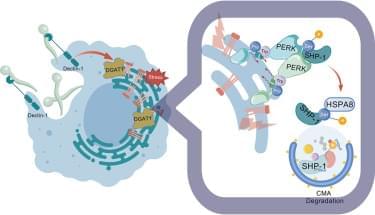
When innate immune cells, such as macrophages, dendritic cells, and neutrophils, encounter fungi, the pattern recognition receptors (PRRs) on their surface recognize evolutionarily conserved fungal cell wall components, including β-glucan and α-mannan (classified as pathogen-associated molecular patterns), thereby initiating downstream signaling cascades and immune responses. The primary PRRs involved in fungal recognition are C-type lectin receptors (CLRs) and Toll-like receptors. The CLR family comprises Dectin-1 (specific for β-glucan), Dectin-2/3 (mannan sensors), Mincle, dendritic cell-specific intercellular adhesion molecule-grabbing nonintegrin, and CD23.5 Upon ligand binding, CLRs initiate the phosphorylation of the immunoreceptor tyrosine-based activation motif within the Dectin-1 cytoplasmic tail and the recruitment of the Fc receptor γ-chain to Dectin-2 or Mincle, which serves as a docking site for spleen tyrosine kinase (SYK).